








发布时间:2019-12-24 点击:次

警示:这些材料所叙述的实验可能是危险的,因此需要高标准的安全训练、特殊的设备和装置,并在合适的人员监管下才能进行。对于履行这样的安全程序和措施,你负有全部的责任和义务,并独自承担其风险。对于所提供的任何材料的内容或其执行情况,MIT将不负任何责任和义务,不承担任何风险。法律提示
目录
1. 简介
1.5. 日程安排..................................................................................................7
1.6. 怎样使用本手册......................................................................................8
1.7. 实验室介绍..............................................................................................9
2. 转移和萃取技术
2.1. CC:“乙酯的轻松之旅”................................................................. 14
2.2. EE:酸、碱及其互变............................................................................17
3. 重结晶纯化固体
3.1. CC:怎样对樟脑球进行重结晶...........................................................19
3.2. EE:单晶培养.........................................................................................21
4. 蒸馏纯化液体
4.1. CC:“桃子是怎样进入香蕉的”...................................................... 24
4.2. EE:高海拔下应该怎么操作................................................................ 26
5. 快速柱色谱纯化法
5.1. CC:“外观可能是欺骗性的”........................................................... 28
5.2. EE:设定速度...........................................................................................30
6. 蛋白质鉴定和误差分析
6.1. CC:“牛的心脏里有什么呢?”....................................................... 32
6.2. EE:“我心坚如磐石”......................................................................... 35
7. 原创性研究介绍
7.1. Mn(salen)配合物催化烯烃的环氧化反应...........................................37
8. 技术指南
8.1. FT-NMR试样制备....................................................................................44
8.2 GC试样制备................................................................................................46
8.3 薄层色谱(TLC).......................................................................................47
8.4. |
萃取和洗涤 |
50 |
8.5. |
无氧操作 |
53 |
8.6. |
双溶剂重结晶 |
55 |
8.7. |
培养单晶 |
57 |
8.8. |
蒸馏 |
59 |
8.9. |
快速柱色谱 |
63 |
9. 仪器操作指南
9.1 NMR............................................................................................................. 68
9.2 IR.................................................................................................................... 73
9.3 GC.................................................................................................................. 74
9.4 UV-Vis.............................................................................................................75
1. 简介
1.1. 概述
欢迎选择课程5.301!本课程特意设计为对实验化学各种技术的集中介绍。本课程的目标有两个:首先,由于本科新生不能选修常规的化学实验室课程,5.301课程的设置可以给有兴趣的一年级学生提供一个动手从事化学实验的训练机会。其次,本课程是为化学系新生的UROP所设立的。新生通常很难在化学系找到一个UROP的工作,因为他们尚未有在正规的化学实验室系列课程中训练出的实验技巧和经验。在接下来的一个月中,你要学习各种基本操作,例如混合、搅拌和测量等,直至你达到化学研究的专业水平。不像其他实验课,我们的目标不仅是成功完成一个实验和写一份实验报告,相反地我们更注重对开展实验所必需的技术和技能的熟练掌握。
在课程5.301中所要学习的技术分为5个不同的模块,每个模块由两部分构成:“基本要求”和“高级要求”,即CC(Competent Chemist)和EE(Expert Experimentalist)。要达到每项技能的CC等级,你须具备一定水平的熟练掌握获得最小数量或最低纯度的某种特定样品的技能。当你的某项技能达到了CC等级后,你可以转入更富挑战性的EE水准的技能训练,那儿有更高的实验要求以证明你达到了EE要求。为了确保你个人的进步,当完成某个模块的训练后,你应当回顾一下每部分开头所列的“实验技能列表”的内容,掂量一下自己能否顺畅地开展这些实验操作。记住,在成为真正的专家之前,你同样可以达到对实验技术非常熟练的程度。我们并没有希望你通过该课程的学习就能在今后的化学研究中对所有的问题能够独立予以解决,你尚需要更多的训练和实践!我们的目标是你能够达到这样一种境界:在遇到不熟悉的问题或技术时,能寻求合适的方法来解决问题。
在本课程的最后一周,将向你介绍一个原创性的研究课题。指导教师将给出一个问题, 由你在实验室中尝试并予以解决。做这个实验,涉及到之前3周内所学的许多实验技术。如果你能正确运用这些实验技术,你一定会向你的指导教师给出一个用实验证明了的解答。
学完了5.301课程后,你将掌握实验室的许多基本操作,并且可以准备迎接更富挑战性的问题。祝你好运!
1.2. 教材
课程5.301选用两本教材:(1)James Zubrick编,有机化学实验室安全手册:学生技能指南,第6版(后面简称Zubrick);(2)J. Leonard,B.Lygo和G.Procter编,高级实用有机化学,第2版(后面简称LLP)。这两本教材之间具有非常好的互补性。虽然他们都是特意为有机化学实验的学生设计的,但书中描述的实验技术也涉及无机化学和金属有机研究。许多实验技术对于生物和物理化学实验同样也是重要的。
Zubrick编写的教材可读性很强,特别适用于刚进入有机化学实验室的学生使用。它包含有相当优秀的实践性建议和很好的解释说明,而且它的内容真的非常有趣。这是一本非常好的学习新的实验技术的启蒙教材。但是,有一点需注意,Zubrick的一些讨论或有些陈旧,或低于课程5.301的要求。因此,我们同时也使用LLP教材。
J.Leonard,B.Lygo和G.Procter编写的教材,同样可读性强,但它是为相对于Zubrick教材居于更高层次的读者而准备的(正如书名所指)。这本教材正好弥补Zubrick的不足,它对实验项目的解释更为详细,相对于本科教学实验室,它对实际的科研实验室有更多的介绍。LLP不仅可以帮你熟悉教学实验室,也可以帮你熟悉科研环境。
1.3. 导读
在1月5日开始进入实验室之前,你必须事先阅读教材中的几个章节。我们在实验室中的时间紧张有限,因此及时完成这些材料的阅读非常重要。这些阅读教材不仅对成功学习课程5.301是必要的,而且可以帮助你成长为一个实验化学家。
因此,在开始做实验之前,先花一些时间来阅读Zubrick中的有关章节。之所以选择该教材,是因为它易读,并且实践性强。要想获得相关主题的更有深度的资料,我们推荐阅读
J.Leonard, B.Lygo和G.Procter编写的教材中的有关章节,在科学图书馆的藏书室里可以找到该书。一般地,仅仅靠简单的教材阅读是难以完全领会一个实验概念的,但是通过运用导读中的策略、实验室中的实践和实验后的复习,你一定会掌握课程5.301中的大部分内容。在每个实验阶段的开始时,都有一个对当天实验的简短讲解,Tabacco博士和助教们还
会组织一个针对所布置的阅读材料和实验室的实际操作的讨论。你也可以通过观看数字化实? 验室技术手册的相关演示,以获得对重要技术的感性认识。同时,会安排时间回答你在预习?中遇到的问题。
下面列出本课程的主要阅读材料。在IAP中,还会有补充阅读材料,该导读内容是希望你能对典型的化学实验室尽快熟悉起来。
我们期待着在1月5日的中午见面!
Zubrick——有机化学实验室安全手册(第6版)
章节:1-安全问题, 2-实验记录,4-接口,6-相关实验器材, 9-清洁和干燥,10-干燥剂,11-产物,15-萃取和洗涤,18-加热,19-夹持器,31-实验室仪表
Leonard, Lygo和Procter-高级实用有机化学(第2版)
章节:1-简介,2-安全问题,3-实验记录,4-实验室装备,8-真空泵
1.4. 等级
概述:
本课程严格以通过和不通过的纪录为基础来确定等级。如果你完成了预定数量的实验,你将获得“通过”。在5.301中,“通过”意味着你有资格在化学研究实验室里开始UROP工作。如果你没有完成所要求的实验,你就不能获得“通过”,并且不能开始UROP工作。然而,有才能的、专注的和有热情的学生不应该觉得难以成功地完成实验的要求。
总共四个星期,你将遇到五个技术模块以及一个原创性研究方案的介绍。前三个星期,训练实验的各项技能,最后一周则用于实践科研课题。每个技术模块中都有两个不同技术难度的练习。如果成功地达到第一个技术水平,便获得“CC?等级”,意味着你在该项技术领域中具备足够的水平,能够完成涉及相关实验技术的研究。如果成功完成每个技术领域的第二部分练习,便赢得“EE等级”,说明你在该项技术领域中具备较高的水平。
要求:
所有技术模块中,无论是“CC等级”还是“EE等级”,都明确给出了要达到该实验相应等级的标准。如果第一次实验没有达到这些标准,你应该重新进行实验,直到达到期望的结果。记住实验化学既是技巧也是一门科学。有些时候,要达到专家的一定水平,你必须进行足够多的实践。
要成功通过课程5.301, 你必须通过所有五个“CC等级”的实验和两个“EE等级” 的挑战。鼓励你完成全部训练,但是仅要求两个“EE等级”的实验。在开展原创性研究课题中,你必须至少完成一个环氧化反应。
1.5.... 5.301日程安排
2004年1月
周日 |
周一 |
周二 |
周三 |
周四 |
周五 |
周六 |
1 |
2 |
3 |
||||
4 |
5 #1 实验室安全;概要; 仪器 |
6 #2 CC: 转移及其操作 |
7 #3 CC: 重结晶 |
8 #4 EE: 转移及其操作或重结晶 |
9 自 选 : 重 做CC或完成EE |
10 |
11 |
12 #5 CC: 蒸馏 |
13 #6 EE: 蒸馏 |
14 #7 CC: 柱色谱 |
15 #8 EE: 蒸馏或 柱色谱(第1天) |
16 自选: 重做CC 或继续柱 色 谱 (第2天) |
17 |
18 |
19 放假 |
20 #9 CC: 生物化学 |
21 #10 EE: 生物化学 |
22 #11 原创性研究:制备 Salen 配体 |
23 #11 原创性研究:制备 Mn 催化剂 |
24 |
25 |
26 # 12 原创性研究:环氧化反应 |
27 #13 原创性研究: 纯化和分析 |
28 #14 原创性研究: 数据整理 |
29 结束实验,整理清点 |
30 上交原创性研究的报告 |
31 |
1.6. 怎样使用本手册
就像整个课程一样,这本手册将向你介绍化学研究的环境。我们几乎不花时间来讨论理论和概念,相反地,着重集中在化学的实践方面。为了便于实践学习,本手册共分为9个部分,这里作一简单介绍。
首先,你正在阅读的“引言”部分,将使你对课程5.301的目标及思想方法有所认识。第二,第2~6部分为实验技术模块,是整个课程的主体。这里包括5个专题:“转移和
萃取”,“结晶纯化”,“蒸馏提纯”,“快速柱色谱纯化法”,“蛋白质鉴定和误差分析”。 必须注意,本手册中并没有涵盖你完成这些实验所需的所有信息。一些重要信息需通过你的实验预习来获得,还有一些将在实验前的讨论中涉及。教材、手册和讨论这三种学习方式相互交叉补充,足以使你能够对付技术模块中列出的实验。
在2~6部分中,一个重要内容便是实验技术清单。每个部分一开始都有一份实验中将遇到的实验技术清单。当你完成一个技术模块时,你应该返回到实验技术列表部分核实一下你已经掌握的所有技能。若你对某项技能还不满意,应该继续练习,直至确信在不同实验中你都能熟练应用它为止。除了各种提纯及操作技术,该部分也向你介绍一些谱学技术,比如: 核磁共振(NMR)波谱,红外(IR)光谱,气相色谱(GC)和紫外可见(UV-Vis)光谱。
第三,第7部分讨论你在IAP的第四周将遇到的原创性研究项目。该部分通过一组实验向你介绍一些实际的、令人兴奋的原创性研究的开展。
第四,第8部分,题为“技术指南”,将一步一步地向你介绍化学实验室中经常遇到的一些通用技术。该指南不仅在5.301中有用,它在你整个研究工作中都是用得上的,因为在研究中会再次遇到这些技术。
最后,第9部分将介绍我们在5.301中所用到仪器的操作方法。这些详细介绍有助于你熟练掌握NMR,GC,IR和UV-Vis等仪器的使用。
1.7. 实验室介绍
在Mircea Gheorghiu博士和Scott Virgil教授的帮助下编成
1. 安全事项
确保你熟悉下列安全设备的放置位置和使用方法:
1. 灭火器,悬挂在实验室中的各个相应位置。
2. 喷淋装置,每个实验室靠近走廊的位置。
3. 洗眼器/?喷脸器,中间通道的每个水槽中有一个。
4. 灭火毯,放在实验室靠近走廊的两头和总电源控制板附近。
5. 电话——仅在紧急情况下使用——拨100。
由化学药品或电引起的火灾,只能用CO2和干粉灭火器进行灭火。水槽处的水龙头可以用来冲洗与腐蚀药品接触的皮肤。你应该注意你所在工作区域中安全设备的放置位置,并且知道(甚至预演)怎样操作,以防火灾或其他的意外事故的发生。但是,在火患或其他意外事件发生的情况下,不要采取任何可能危害到自己或他人的冒险行动。最重要的是,尽快让助教或职员知道所发生的紧急情况。
在实验室,必须一直戴着防护眼镜。这是麻萨诸塞州州法律中的一条,而不仅仅是一条实验室规则。虽然从技术上讲,收音机和乐器并不存在安全隐患,但仍然不允许将他们带进实验室。
学习化学实验室所用到的原料、仪器和操作过程的危险性是本课程教学目标的一部份。星期一(即1月6日)第一次见面时,我们将明确地针对本课程的各种安全注意事项进行讨论。通过讨论,将使你对今后的本科实验中可能遇到的危急情况有心理准备。与此同时,你将拿到一本麻省理工学院化学系的化学保健计划和安全手册,作为你在麻省理工学院生活期间的...安全参考。
溶剂、化学药品及其他材料的处理:
千万不要将溶剂或反应物倒入下水道。对可燃或毒性液体的随意处理是实验室的安全大忌。同时,不能将此类溶剂置于敞口烧杯,放在通风橱外。氯代溶剂应该倒在通风橱内的4-454和4-460废液缸中。当不清楚如何处理某种物质时,请先咨询助教。 如果必需并且可以直接排到下水道时,则必须在排前、期间和排后都用大量水对下水道进行冲洗,而且应该用通风橱中的下水道。用过的玻璃容器必须丢弃在专门设计的容器中。清扫破碎玻璃用的畚箕和扫帚可以从实验供应处(4-450)领取。溢出的水银是非常危险的,应向助教报告,以便进行清除。
2. 登记手续
简单参观了本科生实验室(包括仪器设备和安全设备)之后,便进入实验室登记程序了。将给你指定一个实验位子,并拿到下列物品:
1. 一份安全守则——你必须阅读、签字,然后上交。
2. 实验台和钥匙,仪器清单和登记表。
3. 防护眼镜,实验服和实验记录本(5.301所要求的)。
4. 5.301课程中专用仪器的清单。
比照助教给你的清单,核对实验柜中的仪器。向助教报告你缺失的仪器,助教将会补足你所缺的仪器,或者告诉你去实验室贮藏间领取。一旦在登记表上签字确认,你就得对实验台上所有物品负责。在课程结束时,即便没有通过课程,核对返还所有物品仍然是你的责任
(请见下面的第三条)。
3. 返还仪器程序及收费
2004年1月30日星期四返还实验仪器。没有按期返还仪器的同学名单将由实验供应办公室列出,这部分同学此时需交费$35.00。
4. 实验室仪器的分布
a. 化学药品和溶剂
有机物和无机物——4-457;
酸和碱——4-457的下面厨柜中;
溶剂——4-454和4-460,在实验台末端的架子上。
b. 烘箱和冰箱
烘箱位于4-454和4-460。每个烘箱都有其特定的用途。不要把任何塑料制品放入烘箱。所有样品都要用标签清楚地标明化合物的名称类别、你的名字和日期。烘箱每周清理一次,标识不当的样品将被处理掉。冰箱放在4-454,样品同样需要清楚地加以标识。
c. 天平。不允许滥用天平和破坏其周围整洁。
d. 公用实验室设备
以下物品可由LS提供(LS=Lab Supplies,表示实验室供应处):装上交样品用的瓶子和标签纸;
![]() 滤纸,有17mm、51cm和11cm三种规格;
滤纸,有17mm、51cm和11cm三种规格;
2
橡皮塞、橡胶隔膜和橡皮带;
钳子、针头、锉刀、玻璃管和其它硬件; 海绵、吸油材料、畚箕和扫帚。
5. 麻省理工学院本科化学实验室的安全事项
保护实验室人身健康和安全,保护周围环境是化学系的基本道德规范。一个良好的安全防护计划要求全体教工、职员和学生共同来承担职责。本实验室的安全防护计划由本科生实验室主任Mircea Gheorghiu博士领导,并由一个包括教职员工、助教和学生组成的本科实验室安全委员会负责。
安全信息可通过多种途径获得。每门实验课程都从严格的安全教育开始,同时提供各项相关信息和建议。此外,每门实验的介绍和助教的讲解都有针对具体实验的安全注意事项。实验室中用到的化学品的各种资料都有档案记录,可以到Gheorghiu博士办公室外的资料室借阅。其中,我们特别推荐你阅读《实验室中的细心操作》这本全面的、可读性强的读物。
实验设计和流程要遵守实验室关于有毒物质的规定,有毒物质的使用量要控制在美国政府工业卫生专家会议(简称ACGIH)推荐的阈限值(简称TLV’s)之下。这是一个谨慎的规定,因为这些阈限值能保证一周40个小时连续置身于含有毒物质的工作场合而不会受到危害。ACGIH会议推荐的TLV值在资料室中都能查阅到。
尽管化学系坚决承诺注重本科生实验室安全,但是一定要牢记,绝对无危险的教学环境?是不可能的,也是无法期望的。日常生活中也总有危险存在。比方说,汽油具有爆炸性和毒性,但大多数驾车的市民都相信自己可以安全地使用汽油。任何从事实验科学或医药事业的人都需要学习如何有意识地完善处理各种不同的有潜在危险的物质。本科生实验课程的一个目标就是教会学生如何在实验室内外进行安全操作和实践。
你应该熟悉附录中所列的实验室安全基本守则。同时,你需要熟悉化学保健计划和安全? 手册。严格遵守以上这两份资料上的规则,促使你安全成功地完成实验任务。
6. 本科生实验室的安全总则
1....安全的方法便是正确的工作方法。合理安排工作,遵守实验说明。如果你不知道如何安全地进行实验操作,请向助教咨询。
2....能使用提供给你的所有安全设施和防护设备,清楚它们的放置位置(洗眼器、喷淋装...?置、灭火毯和灭火器)。
3.... 必须始终佩戴安全防护眼镜。
4....不准在实验室进食和饮水(也不可以在冰箱里储存食物)。绝对禁止在实验室吸烟。
5.... 个人防护:穿合适的衣服(处理腐蚀性、毒性或可燃性物质时要穿防护衣)。避免穿有宽松袖子和袖口的衣服,避免带手镯。注意你的长发。穿合适的鞋子(不要穿凉鞋)。
6.... 任何形式的嬉戏打闹都是危险和不允许的。不要在实验区域内跑动。
7.... 如果见到你的同伴有危险举动,向他们指出,并向助教报告。
8....向助教报告所有不安全的情形、不安全的行为和可能引起意外事件的任何状况。无论是多么微小,一定要向助教报告任何的意外事件或火患。
9.... 危险化学药品:
a.当你或你的邻桌在使用可燃液体时,要特别注意火的危险性。
b.危险的物质:了解常见的易爆、有毒和致癌的物质,在有充分的防护措施时才能使用。
10.?绝不能在无人看管的情况下进行化学反应或实验,除非你已经详细地告知你的搭档,在你不在的情况下如何处理潜在的危险情形。
11.?保持通风橱和实验台区域干净整洁,留有最大的工作空间。
2. 转移和萃取技术
2.1. 初级实验要求:“乙酯的轻松之旅”
实验技能列表:
● 萃取和洗涤
● 无损失地仔细转移溶液
● 溶剂蒸发和浓缩
● 熔点测定
● 核磁共振(NMR)波谱仪的操作
实验前的讨论与阅读要求:
● 萃取:Zubrick第15章,LLP第10章
● 萃取理论:Zubrick第37章
● 熔点测定:Zubrick第87-92页
● NMR的理论及操作:Zubrick第35章,LLP第15章第2节
数字化实验室技术手册:
● 5.反应技巧1:萃取、洗涤和干燥
● 6.萃取技巧2:旋转蒸发仪的使用
装置:
● 量筒(100mL)
● 分液漏斗(125mL)
● 锥形瓶(2×250mL)
● 烧杯(150mL)
● 圆底烧瓶(100mL)
● NMR样品管
● 漏斗
● 滤纸
● 旋转蒸发仪
目标:
提纯一定量受污染的样品,记录纯化样品的1H NMR谱图,所有操作中要尽量避免样品的损失。
实验要点:
● 你将拿到一小瓶内含100mg受三乙胺污染的3-羟基苯甲酸乙酯。同时,还有4张不同的1H NMR谱图:一张是小瓶内混合物的谱图,其他三张分别是纯的3-羟基苯甲酸乙酯、三乙胺和乙醚的谱图。

● 在分液漏斗中将样品溶解在50~70mL乙醚中。
● 用10%的HCl溶液萃取,除去三乙胺。
● 继续进行标准的水洗操作,包括用乙醚反相萃取(参见:萃取和洗涤指南)。
● 用旋转蒸发仪中蒸除溶剂至恒重,称重。
● 测定所得样品的1H NMR谱图,并与已知谱图比较。
● 将NMR测试样品和剩余的纯化样品合并。
● 最后一次除溶剂,至恒重。
● 称重,测定熔点。
提示:
● 在用旋转蒸发仪蒸除溶剂时,注意确保接收瓶是在冷却状态,水浴锅是在加热状态。否则你的产品将不会凝固。
● 如果在产品固化时遇到困难,可试着在蒸馏瓶中加入几毫升二氯甲烷,然后再重新进行旋转蒸发。
结果:
● 在转移和萃取技术中,要达到“CC等级”,你至少须获得90mg纯化的3-羟基苯甲酸乙酯。此外,经1H NMR分析,此样品必须足够纯净。这意味着所得的谱图上只允许有可忽略不计的杂质峰,并由教授或助教判断认可。另外,纯化后产品的熔程不超过
3℃,下限不低于69℃,上限不高于73℃。产品需交给助教,以验证所得样品的重量及熔点。
2.2. 高级实验要求:“酸、碱及其互变”
实验技能列表:
● 根据pKa分离多组分混合物
● 设计萃取和洗涤程序
● 无损失地仔细转移溶液
● 溶剂蒸发和浓缩
● 熔点测定
实验前的讨论与阅读要求:
● 与“CC”一样
数字化实验室技术手册:
● 与“CC”一样
装置:
● 量筒(100mL)
● 分液漏斗(125mL)
● 锥形瓶(4×250mL)
● 烧杯(150mL)
● 圆底烧瓶(100mL)
● pH试纸
● NMR样品管
● 漏斗
● 滤纸
● 旋转蒸发仪
目标:损失最少的原则下,根据pKa的差别,分离三组分混合物。
实验要点:你将拿到一小瓶含有苯甲酸、4-硝基苯胺和萘共100mg的样品。根据pKa值, 仔细设计萃取和洗涤程序,选择性分离三个组分1。
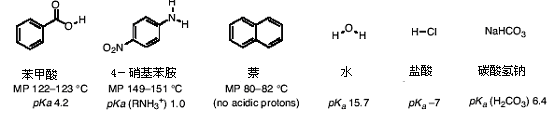
● 在开始萃取实验前,请助教或教授检查,以确保你的方案可行。
● 你可以随意选用下列溶剂和溶液:
—乙醚
—甲醇
—水
—饱和Na2CO3水溶液
—6M HCl
—1M NaOH
● 进行萃取和洗涤,分别分离出三个组分。
● 对每一个化合物,在旋转蒸发仪中蒸掉溶剂至恒重,称重。
● 测定每一个化合物的熔点。
结果:
● 在转移和萃取技术中,要达到“EE等级”,你必须分离出三元混合物中的两种化合物至少90mg。另外,分离后的化合物的熔程不得超过3℃,最低熔点不低于文献值2℃。
——————————
1选自Gilbert J. C; Martin S F.Experimental Organic Chemistry: A Miniscale & Microscale Approach; 3th ed.; Brooks/Cole: Pacific Grove, CA 93950; p. 141.
3. 重结晶纯化固体
3.1. 初级实验要求:“怎样对樟脑球进行重结晶?”
实验技能列表:
● 溶解度试验
● 优良溶剂体系的选择
● 诱导结晶
● 过滤
实验前的讨论与阅读要求:
● 重结晶理论:Zubrick第13章,LLP第11.2节。
数字化实验室技术手册:
● 9.?重结晶
装置:
● 试管(5只,13×100 mm)
● 锥形瓶(2×50-mL, 1×125-mL)
● 小号磁力搅拌子
● 漏斗
● 滤纸
● 布氏漏斗和滤纸
● 磁力搅拌/加热器
● 抽滤瓶(250mL)和夹子
● 橡皮过滤接头
● 带白盖的大号瓶
● 试管架
● 大号结晶皿
● 干燥器
目标:
你将拿到2.00g不纯的萘(即樟脑球!)。你的任务是在损失尽可能少的情况下对萘进行重结晶纯化!1
实验要点:...........................................................................................萘
I. 溶解度试验
● 为萘的重结晶确定合适的溶剂体系。可以试验如下溶剂:水,甲醇,丙酮,正己烷和甲苯。如何选择重结晶所需的合适溶剂或混合溶剂,请参考Zubrick第104-105页。
II. 萘的重结晶
● 将原料装入50mL带搅拌磁子的锥形瓶中。加入20mL左右的溶剂(通过步骤I确定),在带加热的磁力搅拌器上加热至沸腾。
● 过滤除去不溶性杂质,使产品重结晶析出(参见:二元溶剂重结晶指南)。
● 在小的布氏漏斗中减压抽滤,收集晶体,并用冷的混合溶剂洗涤。
● 所得的晶体应该是无色的。若晶体呈橙色或黄色,应该用冷的正己烷继续洗涤。
(注意:萘在正己烷中的溶解度是多少?)
● 充分干燥所得产品(参见:二元溶剂重结晶指南)。
● 计算回收率,测量熔点。
结果:
● 要在重结晶纯化固体操作中获得“CC等级”,你应该获得至少1.30g无色(没有任何黄色的痕迹!)干燥的晶体。纯化产品的熔程不超过3℃,且熔点最低不低于77℃,最高不超过83℃。该产品需交给助教,以验证所得样品的重量及熔点。
1选自Fieser, L. F.; Williamson, K. L.Organic Experiments;7th ed.; D. C. Heath and Company: Lexington, MA, 1992; p. 40.
3.2. 高级实验要求:“单晶培养”
综述:
X-射线衍射是测定固体化合物结构的一种重要而有效的工具。现代技术的发展已经使得许多小分子的数据采集和结构解析成了日常工作。然而,高质量的单晶体在使用该技术时仍然是必须的。在本实验中,你将体验培养单晶艺术。
实验技能列表:
● 毫克级样品的操作
● 注射器的使用
● 培养高质量单晶的结晶技术
实验前的讨论:
● 不同重结晶技术的应用:蒸汽扩散法,溶剂分层法和温度变化法
目标:
● 合成Cr(acac)3,2然后通过几次缓慢重结晶,获得满意的单晶。
装置:
● 磁力搅拌器
● 电热套和调压器
● 50mL圆底烧瓶
● 2mL玻璃注射器
● 冷凝器
● 搅拌磁子
● 玻璃过滤器(D)
● 250mL抽滤瓶和橡皮过滤接头
● 瓶子(3只大号、4只小号)
● 2只玻璃罩
乙酰丙酮
![]()
2选 自 :Szafran, Z.; Pike, R. M.; Sing, M. M. Microscale Inorganic Chemistry: A Comprehensive Laboratory Experience, Wiley: New York, 1991: “synthesis of Metal Acetylacetonates” p. 224-229.
实验要点:
● 实验前,进行必要的计算,并填写下表:
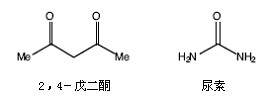
试剂 |
来源 |
分子量 |
密度 |
质量或体积 |
毫摩尔数 |
当量 |
CrCl3·6H2O |
1.00mmol |
1 |
||||
尿素 |
17 |
|||||
2,4-戊二酮 |
8 |
|||||
Cr(acac)3 |
产品 |
●
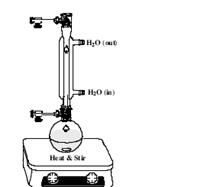
在配有搅拌磁子的50mL圆底烧瓶中,将CrCl3·6H2O溶于2mL蒸馏水中。
● 将尿素一次加入到烧瓶中,搅拌,直至完全溶 解。
● 将2,4-戊二酮通过注射器逐滴加入。
● 在烧瓶上装上冷凝器,在搅拌下加热混合物使其剧烈回流(非常重要!)约1小时。
● 冷却反应液至室温,用D型玻璃漏斗减压抽滤产 品,用冷水洗涤产品。
● 将产品置于干燥器中干燥过夜,计算产率。
● 通过多次重结晶,培养出适合X-射线衍射分析用的单晶
(参见:单晶培养指南)。
注意:
反应装置
● 尿素在反应条件下会缓慢水解,释放出控制反应pH值的氨气(NH3)。产生的
NH3越多,溶液的碱性就越强,使得乙酰丙酮(acac)(即2,4-戊二酮)上的质子更容易离去,对2,4-戊二酮也是如此。得到的乙酰丙酮阴离子可以和金属配位形成目标配合物Cr(acac)3。最终产物是什么?请计算产率。
提示:
● 当用饱和溶液来生长晶体时,在重结晶前,通过玻璃毛过滤溶液是非常重要的。
结果:
● 要想达到“EE等级”,你必须获得大于50%产率的Cr(acac)3,并且至少培养出一个适合于X-射线衍射分析的单晶。
4. 蒸馏纯化液体
4.1. 初级实验要求:“桃子是怎样进入香蕉的?”
实验技能列表:
● 正确组装玻璃蒸馏装置
● 常压蒸馏的操作
● 用气相色谱(GC)分析样品
实验前的讨论与阅读要求:
● 蒸馏理论:Zubrick第36章,LLP第11章第3节
● 蒸馏用玻璃仪器及其组装:Zubrick第20章
● GC的使用:Zubrick第32章
装置:
● 圆底烧瓶(1×25mL, 1×50mL)
● 搅拌磁子
● 蒸馏头
● 磨砂玻璃温度计
● 夹子
● 玻璃毛和铝箔(任选)
● 加热套及调压器
目标:
● 用蒸馏法提纯二元液体混合物。
实验要点:
● 你将拿到一瓶装有11.20g的二元液体混合物,两组分的沸点相差约40℃(下面给出了可能化合物的结构)。
● 用GC分析混合物(参见:气相色谱样品的制备与操作指南)。
● 进行常压蒸馏操作(参见:蒸馏操作指南)。
● 用你提纯得到的低沸点产物准备一个GC试样。
● 称量纯化所得的低沸点物质的质量,并测定其气相色谱图。
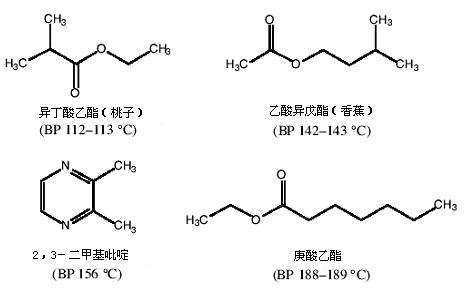
提示:
● 确保装置的各接口处密封良好。否则你的产品将会流失到空气中。
● 用棉花或铝泊包裹蒸馏头,以加快蒸馏速度。
● 加热速度不要太快,否则所有样品将会一起冲入到接收瓶中。
● 注意温度计上读数可能与馏出液的沸点不十分吻合。
结果:
● 要想在蒸馏纯化液体的实验技术中获得“CC等级”,你必须至少获得7.00g的低沸点物质,其GC分析纯度应等于或超过92%。你必须能正确地鉴别混合物中的两个组分。可以借助沸点和气味喔!
4.2. 高级实验要求:“高海拔下应该怎么进行操作?”
实验技能列表:
● 减压蒸馏装置的组装
● 减压蒸馏操作
实验前的讨论:
● 常压蒸馏与减压蒸馏的区别
装置:
● 25mL圆底烧瓶
● 减压蒸馏组件(蒸馏头,多头接口)
● 接收瓶
● 磨砂玻璃温度计
● 夹子
● 玻璃毛和铝箔
● 加热套及调压器
目标:
● 通过减压蒸馏,提纯二元液体混合物。
实验要点:
● 你将拿到一小瓶有α-紫罗兰酮和十八烷组成的混合物7.50g。
● 用维氏(Vigreux)分馏柱和高真空装置,重复“CC等级”的蒸馏操作。(参见:
蒸馏操作指南)。
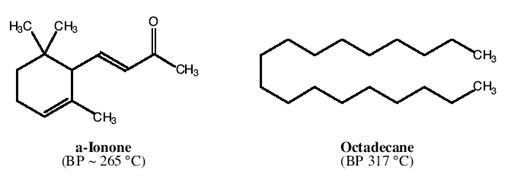
结果:
● 要想在蒸馏提纯液体实验中获得“EE等级”,你必须会预测混合物中每种组分在0.5torr压力下的沸点。同时,获得至少4.00g α-紫罗兰酮,而且气相色谱分析纯度大于等于93%。
下图是一个列线图。用这张图和一把尺子,你便可以确定减压下液体在什么温度沸腾。
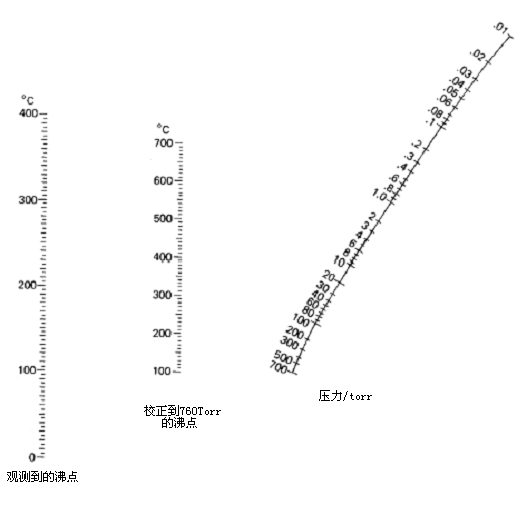
5. 快速柱色谱纯化法
5.1. 初级实验要求:“外观可能是欺骗性的”
实验技能列表:
● 用TLC分析混合物
● 硅胶柱的装填
● 在硅胶柱中添加粗产品混合物
● 用硅胶柱分离简单的混合物
实验前的讨论与阅读要求:
● 柱色谱的原理:Zubrick第27章
●TLC-极性/溶剂体系:Zubrick第28章,LLP第9.3.1节
● 硅胶柱的制备:Zubrick第29章,LLP第11.6节
● 柱中粗产品混合物的添加
● 快速层析柱的操作
装置:
● 快速色谱柱
● 气体流动装置(塞子,T型阀,螺旋夹,管道)
●100mL圆底烧瓶
● 试管18×150 mm
● 试管架
● 薄层色谱板和点样器
● 紫外灯
● 大号塑料漏斗
数字化实验室技术手册:
●3. TLC:基本原理
●10.?柱色谱
目标:
● 用硅胶快速柱色谱方法提纯一个受污染的化合物。
实验要点:
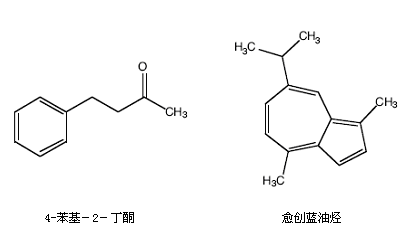
● 你将拿到2mL?受少量愈创蓝油烃(guaiazulene)污染的4-苯基-2-丁酮
(benzylacetone)的乙醚/戊烷溶液。
● 制备一个TLC分析样品,并以10%乙酸乙酯/正己烷为溶剂进行TLC分析。
(参见:TLC指南)
● 记录Rf值。
●用10%乙醚/正戊烷混合溶剂和50g(大约是5目)的硅胶,在通风橱中制备色谱柱。(参见:快速柱色谱法指南)
● 用10mL正戊烷进行淋洗。
●将样品加入柱中,注意不要扬起柱顶部的沙层。分别用1mL正戊烷冲洗试样瓶三次,并用此清洗液淋洗柱壁。
● 过柱。(参见:快速柱色谱法指南)
● 用TLC分析洗脱馏分。(参见:薄层色谱指南)
● 浓缩含有纯4-苯基-2-丁酮(benzylacetone)的馏分。
● 称量所得的产物,并准备GC分析所用的试样。
● 测试纯样品的TLC和气相色谱图。
结果:
● 要想在快速色谱柱纯化法实验技术中获得“CC?等级”,你必须能收集到至少
0.95g的纯4-苯基-2-丁酮,且样品的气相色谱纯度至少达到95%以上。请将产品交给助教,以验证其重量和纯度。
5.2. 高级实验要求:“设定速度”
***...?这个实验需要两天时间。请确认你有连续两天时间可自由支配时再开始实验。***实验技能列表:
● 选取合适的洗脱液
● 在硅胶上吸附粗产品混合物
● 用梯度洗脱法分离复杂混合物
实验前的讨论:
● 列出几种洗脱溶剂体系的清单
● 讨论样品吸附和梯度洗脱的程序
装置:
● 和“CC”级相同
目标:
● 用梯度淋洗快速柱色谱分离由三种化合物组成的复杂混合物。
实验要点:
●你将拿到20mL内含0.2g愈创蓝油烃(guaiazulene)、0.2g芴酮(fluorenone)以及0.2g3-甲基苯甲醚的乙醚/戊烷混合溶液。
● 用预讨论中确定的溶剂体系对混合物进行TLC分析。(参见:TLC指南)
● 确定最先使用的洗脱液。
● 确定硅胶与化合物的组成比。
● 制备色谱柱。(参见:快速柱色谱法指南)
● 根据实验前的讲解和数字化实验室技术手册中提供的材料,将样品吸附到少量的硅胶上。
● 将混合物装入色谱柱,注意要对柱壁进行冲洗,并在柱顶再铺上一层沙。
● 过柱。
●用TLC分析洗脱的所有组分,将你的薄层层析板给指导老师检查,并将层析板画在实验记录本上。
● 计算三个化合物在所选定的TLC混合溶剂中的Rf值。
结果:
●要在快速柱色谱纯化技术实验中获得“EE等级”",你必需用薄板层析(TLC)成功分离出混合物中的所有组分,并正确地计算出每种物质的Rf值。
6. 蛋白质鉴定和误差分析
6.1. 初级实验要求:“牛的心脏里有什么呢?”
实验技能列表:
● 用吸移管定量移液
● 吸移管校正
● 标准曲线制作
● 梯度稀释
● 紫外-可见(UV-Vis)分光光度计使用
实验预讨论:
● 蛋白质鉴定
装置:
● 吸移管:100P,1000P
● 洗耳球:大、小型
● 8支试管
● Eppendorf管和管架
● UV-Vis一次性比色皿(5mL)
目标:
●你将拿到含有牛心细胞色素C的溶液样品,并且用Pierce公司的Coomassie? Plus Protein Assay试剂来鉴定样品中蛋白质的浓度。
注意:
● 你将拿到一组Eppendorf管:一个装储备液,3个装有50μL的牛心细胞色素C,以及若干用于混合溶液的空管。还有一瓶pH为7的25mM MOPS缓冲溶液。
实验要点:
吸移管的校正
在实验中使用吸移管以前,首先应对它进行校正。该步骤将决定移入液体体积的精确值。对移液管的校正非常方便,只需吸取一定量的水,然后将它们移入一个已知重量的带盖容器,然后再称取其重量。由于已知水的密度是1.00g/mL,因此,进行简单的换算就可以知道吸移管的准确度。大多数吸移管不需要修正,若要修正,一般不应该超过1μL。
Coomassie?试剂和蛋白质的反应
蛋白质的检验可以通过其和Coomassie?染料形成的配合物来实现。当染料附着在蛋白质上之后,染料的紫外吸收会从465nm移至595nm(A595)。首先测定一系列已知浓度的牛血清蛋白(BSA)溶液在595nm处的吸光度,绘制标准曲线,然后测定未知样品在595nm处的吸光度,对比标准曲线确定样品的浓度。
蛋白质+酸性介质中G-250 Coomassie?→蛋白质-染料配合物(蓝色,可在595nm下检测)
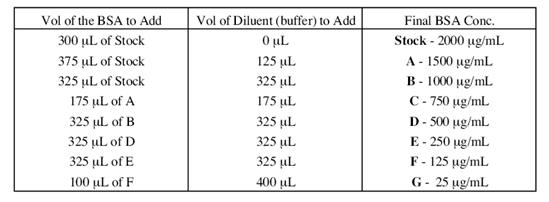
1.配置稀的BSA标准溶液
● 将2.0mg/mL BSA标准溶液的储备液按下表稀释,配置一组新鲜的蛋白质标准溶液。如果需要的话,每一稀释的BSA标准溶液体积足以满足3次重复实验。
2.Coomassie?和蛋白质检测试剂的混合
● 使Coomassie? Plus试剂恢复到室温。在使用前夕,把Coomassie? Plus和蛋白质检测试剂混合,轻轻翻转瓶子几次即可,切忌振荡。
3.标准曲线绘制
● 吸取0.05mL每一标准溶液到标记好的Eppendorf管中。准备至少3个未知的溶液样品。用0.05mL稀释剂(25mM MOPS缓冲溶液,pH为7,由助教提供)制备两个参比溶液。
● 在每个管内加入1.5mL Coomassie? Plus试剂,混合均匀。
● 用水作参比,测定每个管内溶液在595nm处的吸光度。
● 将标准样品和未知样品在595nm下的读数扣除参比样品在595nm下的读数。
● 绘制595nm下的标准曲线,以扣除空白值的标准样品的吸光度平均值作为纵坐标,其浓度(μg/mL)为横坐标作图。以此标准曲线确定每个未知样品的蛋白质浓度。
提示:
● 要将所有溶液保存至标准曲线绘制和数据分析完毕,因为你有可能需要重测部分样品的紫外吸收。
结果:
● 要在蛋白质鉴定和误差分析实验中获得“CC?等级”,你所绘制的标准曲线的线性相关系数(R)必须在0.930及以上。你所得到的未知样品的吸光度值的标准偏差必须小于在0.048。最后,你还必须确定未知蛋白质溶液的浓度。
6.2. 高级实验要求:“我心坚如磐石”
实验技能列表:
● 离心机的使用
装置:
● 一次性UV-Vis比色皿(1mL)
● 吸移管:20P,100P,1000P
● 洗耳球
● Eppendorf管(带安全扣)
● 加热板
● 离心机
● 加热盘或Eppendorf管架
● 大号结晶皿
目标:
● 在“CC”实验中,你已经测定了样品中蛋白质的浓度。在本实验中,你将测定牛心细胞色素C中铁的含量。
实验要点:
用铁试剂(Ferrozine)鉴定
铁试剂是一种铁的螯合剂,当它与二价铁离子(FeⅡ)形成配合物时,在紫外-可见
(UV-Vis)的562nm处具有特征吸收。通过比较你的样品和铁离子标准曲线的A562,便可以得知蛋白质样品中铁的含量。
由助教提供的溶液:
-Fe的AA标准溶液(AA为原子吸收)
-缓冲溶液(25mM MOPS,PH为7)
-超纯HNO3(5 M)
-75 mM维生素C酸溶液
-10 mM铁试剂溶液
-饱和的乙酸铵溶液
1. 准备标准样品:
●在2mL的Eppendorf管中配制一系列铁的标准溶液,具体如下表所示。注意每个管都要贴上标签,作好标记。同时,在2个管内加入300μL的蛋白质样品。

● 向标准溶液和待测样品中加入30μL超纯的硝酸溶液(5M)。
●塞好塞子,将Eppendorf管放置在管架上,在热水浴(由加热板上放一大的Pyrex
耐热玻璃皿构成)上加热30分钟。
● 离心分离1~2分钟,注意保持离心机的平衡。
●从每个管内移出300μL的上层清液,转移到另外的干净管内(注意做好标记!)。
● 加入1020μL蒸馏水。
● 加入60μL 75mM的维生素C酸溶液。
● 加入60μL 10mM的铁试剂。
● 加入60μL饱和乙酸铵溶液。
● 摇动管子并等待10~15分钟(此时溶液应该变成紫色)。
●将其转移到1.5mL的比色皿中,在562nm处,测量标样和两个待测样品的吸光度
A562。
● 以标样的A562对[Fe]作图绘制标准曲线。
● 测定样品中的[Fe]。
结果:
● 要在蛋白质鉴定和误差分析实验中获得“EE等级”",你所制作的标准曲线的线性相关系数必须达到0.995及以上。另外,你所得到的未知样品的吸光度值的标准偏差必须在0.035之内。最后,必须确定每个蛋白质分子中铁的数目。
7.原创性研究介绍
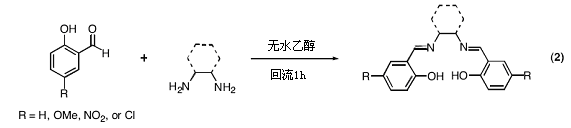
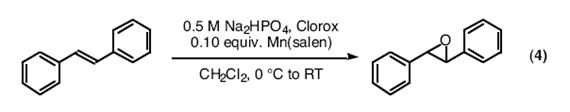
7.1. Mn(salen)配合物催化的烯烃的环氧化反应
引言:
在课程5.301中,你的指导教师已经仔细观察到了你的实验技能的进步。并且确信你可以从技术演练转入实际操作的环节了。这个环节将需要你运用在过去三个星期里学会的各种实验技巧来解决某个特定的问题。此外,你和你的实验伙伴将学习如何以研究团队进行工作, 以在较短的时间内达到一个雄心勃勃的目标。
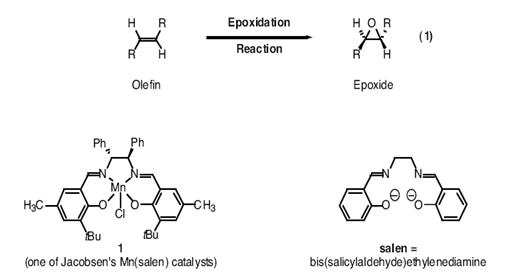
综述(导师的话):
我们课题组对烯烃的环氧化反应(eq1)一直都非常关注,现在想从另一个角度来审视十几年来报道的这个重要成果。Eric Jacobsen(以前是麻省理工学院的博士后,现在在哈佛任教授)指出,图1所示的锰配合物Mn(salen)是非常有效的环氧化反应催化剂(eq 1)。由于锰配合物Mn(salen)容易制备,因此我们想要研究一系列不同的Mn(salen)衍生物的相对催化活性。本实验室的一个研究生已经考察了几种锰配合物Mn(salen),但我希望你和你的实验伙伴能进行更加全面的深入研究。因此,请先阅读所附的Jacobsen的文章,再与研究生讨论课题中即将进行的反应,并且尽力和你的实验伙伴相互配合,最后告知我你们有怎样的思路。
目标:
● 合成一种新的salen配合物(eq 2),并测定1H NMR谱。
● 合成相应的锰配合物Mn(salen) (eq 3)。
● 和你的实验伙伴比较一下催化剂的分离产率。
● 采用Jacobsen报道的实验方法和你制备的催化剂,将1,2-二苯乙烯环氧化。
● 通过1H NMR分析,确定反应的转化率。
● 用快柱色谱纯化粗产品,并计算产量。
● 测定纯环氧化反应产物的1H NMR谱。
● 和实验伙伴比较各自反应的转化率和分离产率。
● 用你的实验结果预测一下催化剂结构和催化效率之间的关系。这有助于今后设计出更好的催化剂。
来自研究生学长的提示:
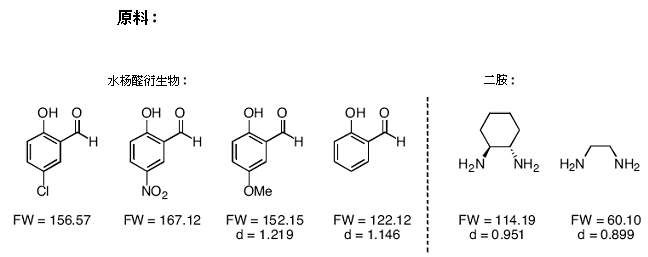
1.制备Salen配体(eq 2)
● 一些你不必尝试的反应:(我在用下列配体制备Mn(salen)配合物时遇到了问题)
1) 乙二胺+水杨醛
2) 反式-1,2-二氨基环己烷+3,5-二-叔丁基-2-羟基苯甲醛
3) 乙二胺+硝基水杨醛
4) 乙二胺+氯代水杨醛
● 你应设法得到1.00克Mn(salen)配合物。若产率可超过70%,计算一下两个反应的投料量。
● 由于反应将会产生大量固体,需要用较大的搅拌棒进行快速搅拌。
● 不要忘记,在反应结束后你需要加水沉淀产物,因此反应容器不能太小。
● 配体(以及之后的金属配合物)刚分离出来时非常潮湿,应先放置在干燥器中过夜, 干燥后再行称重。
● 注意乙醇和无水乙醇的区别。
● 有些我们的salen配体和Jacobsen所报道的有所不同。比如,在有的情况下,需在热酒精溶液中加入甲苯来溶解配体。
●
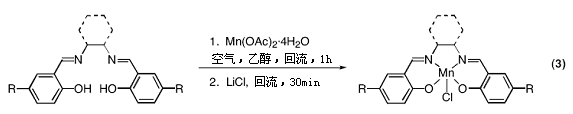
反应完全后,冷却溶液促进Mn(salen)配合物结晶是至关重要的。将反应瓶直接放在冰水浴中冷却约一个小时,再过滤即可。如果在冰水浴中没有固体析出,则可将溶液放在冰箱中冷却过夜。
3. 1,2-二苯乙烯的环氧化反应(eq 4)
● 用pH计测量该反应准确的pH值是最好的方法。当需要用到的时候,助教们会给你演示如何使用pH计。
● 由于有机层位于容器底部,用薄层层析(TLC)来跟踪该反应可能得不到真实结果。最好的方法是将一个吸移管尽可能深地插入混合液。通过毛细作用使液体进入吸移管中,再用正戊烷可将其洗入一个小瓶中。这样,低密度的有机层就会处于水相上层,便于薄层层析点样。注意:我们之所以使用这种繁琐的稀释方法,是因为该反应在二氯甲烷中呈两相。然而大多数的均相反应溶液(或低密度有机溶剂的两相混合物)都可以直接用于薄板点样。
8.1. FT-NMR试样制备指南
综述:
一个好的1H NMR样品约含有10mg左右化合物。样品溶液中不应存在固体或顺磁性的杂质。氘代NMR溶剂中不能含有水分,测得的NMR谱图中不应出现溶剂峰。
参考资料:
Zubrick第295页的内容对你很有帮助,但是你还必需遵守该指南中的详细说明。另外, 请记住:我们不可能一直在波谱仪上进行操作,因此,获得谱图后先继续实验,不要讨论。
NMR溶剂:
典型的氘代溶剂有氘代氯仿(CDCl3),重水(D2O), 氘代苯(C6D6), 氘代丙酮
(CD3C(O)CD3),氘代乙腈(CD3CN)和氘代四氢呋喃(C4D8O)。氘代氯仿是至今最为常用的溶剂,将在5.301中被指定使用。本课程中使用的CDCl3由助教准备。将来你买到瓶装CDCl3时, 需要你自己事先准备。在CDCl3可用作NMR熔剂之前,必须完成三个重要的步骤:一、在
CDCl3中加入数滴标准物TMS(四甲基硅烷);二、加入经活化的4?活性分子筛脱水,确保溶剂中没有剩余的水;三、必要的话,加入无水K2CO3颗粒(一种弱碱),以中和CDCl3和分子筛带来的酸性。在5.301中,我们使用的CDCl3已经经过分子筛的处理,并已经加了TMS, 由于我们不会用到对酸敏感的物质,所以没有加入K2CO3。(注意:不要让水进入氯仿,尽量缩短盛氯仿的瓶子敞开的时间。只要瓶子敞开,空气中的水就会溶进NMR溶剂中。)
在准备试样之前:
1) 检查NMR样品池中的深度测量器,确定所需样品的最低高度。
2) 做一次标准测试,以确定你的待测样品中含有足够的溶剂。(提示:将NMR管放在
10mL的量筒中,在量筒的外面用记号笔在最低检测高度处做上标记。
准备液体NMR样品:
1) 干燥,去除化合物中的溶剂。
2) 取一支洁净干燥的NMR管,放置于10mL的量筒中。
3) 在NMR管的顶部放置一个Kimwipe吸管过滤器。这是通过把一小片Kimwipe填充到一支小的Pasteur吸管中做成的。用大的Pasteur吸管的尖端可以把它塞进相应部位。
(这个过滤器可以除去任何不溶性杂质。)
4) 另取一支吸管,将尖端插入样品中,毛细管效应可使约10毫克样品进入管中。
5) 将吸管插入吸管过滤器,用氘代溶剂将样品洗入NMR管中。
6) 检查溶剂是否足够。
7) 盖上NMR管的盖子,如果需要完成多张谱图,请记录样品号。(在彩色的NMR盖上直接书写编号是最方便的)
8) NMR测试完毕后,将样品回收到原来的容器中,浓缩除去溶剂。
准备固体NMR样品:
1)重复以上的1-3步骤。
2)将10mg左右的样品放在一个小瓶中。
3)用1mL左右的氘代试剂溶解样品。
4)用吸管将液体通过吸管过滤器移入NMR管。
5)重复以上的6-8步骤。
清洗NMR管:
1)用丙酮彻底清洗NMR管。
2)把NMR管放入烘箱中,干燥1小时左右。
3)将NMR管放入室温下的干燥器中保存。
8.2.气相色谱(GC)试样制备指南
综述:
该指南描述如何制备一个标准的气相色谱(GC)检测试样。内容包括挥发性化合物稀溶液的制备和使用气相色谱检测其纯度。
参考文献:
Zubrick 252页。
液体样品制备:
1) 将Pasteur吸管的尖端插入液体中,毛细管现象将使约10mg的液体进入吸管中。
2) 用1mL挥发性溶剂(乙醚、乙酸乙酯和戊烷等)将其洗入一个小瓶中。
3) 将吸管的尖端插入液体中。
4) 用1mL相同的溶剂将吸管中的试样通过吸管过滤器洗入另一个小瓶中。
5) 现在你的样品可以直接进样了!
固体样品制备:
1) 将10mg的化合物溶入1mL上述任一种挥发性溶剂中。
2) 重复以上步骤的3-5步。
8.3. 薄层色谱(TLC)的使用指南
综述:
薄层色谱(TLC)是一种非常有用的跟踪反应的手段,还可以用于柱色谱分离中合适溶剂的选择。薄层色谱常用的固定相有氧化铝或硅胶,它们是极性很大(标准)或者是非极性的(反相)。流动相则是一种极性待选的溶剂。在5.301中以及大多数实验室实验中,都将使用标准硅胶板。将溶液中的反应混合物点在薄板上,然后利用毛细作用使溶剂(或混合溶剂)沿板向上移动进行展开。根据混合物中组分的极性,不同化合物将会在薄板上移动不同的距离。极性强的化合物会“粘”在极性的硅胶上,在薄板上移动的距离比较短。而非极性的物质将会在流动的溶剂相中保留较长的时间从而在板上移动较大的距离。化合物移动的距离大小用Rf值来表达。这是一个位于0~1之间的数值,它的定义为:化合物距离基线(最先点样时已经确定)的距离除以溶剂的前锋距离基线的距离。
参考资料:
参考LLP145-152页,那里有比较全面的讨论。
薄层色谱(TLC)实验步骤:
1)切割薄板。通常,买来的硅胶板都是方形的玻璃板,必需用钻石头玻璃刀按照模板的形状进行切割。在切割玻璃之前,用尺子和铅笔在薄板的硅胶面上轻轻地标出基线的位置(注意不要损坏硅胶面)。借助锋利的玻璃切割刀和一把引导尺,你便可方便地进行玻璃切割。当整块玻璃被切割后,你就可以进一步将其分成若干独立的小块了。(开始的时候,也许你会感到有一些难度,但经过一些训练以后,你便会熟练地掌握该项技术。)
2) 选取合适的溶剂体系。化合物在薄板上移动距离的多少取决于所选取的溶剂不同。在戊烷和己烷等非极性溶剂中,大多数极性物质不会移动,但是非极性化合物会在薄板上移动一定距离。相反,极性溶剂通常会将非极性的化合物推到溶剂的前段而将极性化合物推离基线。一个好的溶剂体系应该使混合物中所有的化合物都离开基线,但并不使所有化合物都到达溶剂前端,Rf值最好在0.15~0.85之间。虽然这个条件不一
定都能满足,但这应该作为薄层色谱分析的目标(在柱色谱中,合适的溶剂应该满足
Rf在0.2~0.3之间)。那么,应该选取哪些溶剂呢?一些标准溶剂和他们的相对极性(从
LLP中摘录)列于如下:
强极性溶剂:
甲醇〉乙醇〉异丙醇中等极性溶剂:
乙氰〉乙酸乙酯〉氯仿〉二氯甲烷〉乙醚〉甲苯非极性溶剂:
环己烷,石油醚,己烷,戊烷常用混合溶剂:
乙酸乙酯/己烷:常用浓度0~30%。但有时较难在旋转蒸发仪上完全除去溶剂。乙醚/戊烷体系:浓度为0~40%的比较常用。在旋转蒸发器上非常容易除去。
乙醇/己烷或戊烷:对强极性化合物5~30%比较合适。
二氯甲烷/己烷或戊烷:5~30%,当其他混合溶剂失败时可以考虑使用。
3) 将1~2mL选定的溶剂体系倒入展开池中,在展开池中放置一大块滤纸。
4)将化合物在标记过的基线处进行点样。我们用的点样器是买来的,此外,点样器也可从加热过的Pasteur吸管上拔下(你可以参照UROP)。在跟踪反应进行时,一定要点上起始反应物、反应混合物以及两者的混合物。
5) 展开:让溶剂向上展开约90%的薄板长度。
6)从展开池中取出薄板并且马上用铅笔标注出溶剂到达的前沿位置。根据这个计算Rf的数值。
7) 让薄板上的溶剂挥发掉。
8)用非破坏性技术观察薄板。最好的非破坏性方法就是用紫外灯进行观察。将薄板放在紫外灯下,用铅笔标出所有有紫外活性的点。尽管在5.301中不用这种方法,但我们将采用另一常用的无损方法——用碘染色法。(你可以参看UROP)。
9)用破坏性方式观测薄板。当化合物没有紫外活性的时候,只能采用这种方法。在5.301中,提供了很多非常有用的染色剂。使用染色剂时,将干燥的薄板用镊子夹起并放入染色剂中,确保从基线到溶剂前沿都被浸没。用纸巾擦干薄板的背面。将薄板放在加热板上观察斑点的变化。在斑点变得可见而且背景颜色未能遮盖住斑点之前,将薄板从加热板上取下。
10)根据初始薄层色谱结果修改溶剂体系的选择。如果想让Rf变得更大一些,可使溶剂体系极性更强些;如果想让Rf变小,就应该使溶剂体系的极性减小些。如果在薄板上点样变成了条纹状而不是一个圆圈状,那么你的样品浓度可能太高了。稀释样品后再进行一次薄板层析,如果还是不能奏效,就应该考虑换一种溶剂体系。
11) 做好TLC标记,计算每个斑点的Rf值,并且在笔记本中画出图样。
8.4. 萃取和洗涤指南
综述:
该指南描述标准的萃取和洗涤方案,此方案可用于任何反应的粗产品混合物。你的预期产物通常溶于有机层,因此通过水洗可以从有机产品中除去水溶性的杂质。
参考资料:
更完整的讨论,请参阅:Zubrick第127-138页。
标准水洗方案:
1)选择有机溶剂。乙醚是最常用的有机溶剂,因为可方便地用旋转蒸发仪将其除去。乙酸乙酯也是很好的溶剂,但是它相对比较难被除去。应该尽量避免使用二氯甲烷, 因为二氯甲烷比水重,容易形成难以处理的乳状液和复杂的物质。
2)选择分液漏斗的大小。通常选用125mL或250mL的分液漏斗,较大量的反应(1~
10g)可以用500mL或1L的分液漏斗。请记住:分液漏斗中要装得下溶剂及洗涤液,两者在漏斗中必须能完全混合。
3)用所选择的有机溶剂稀释初始反应混合物并将其移入选择好的分液漏斗。大量的原?料需要大量的溶剂。常规反应(50~500mg产品)可用25~100mL溶剂来稀释。
4)洗涤有机层以除去杂质。洗涤相的体积通常是有机相体积的1/10~1/2。最好重复洗涤2~3次。酸洗(通常用10%HCl)可以除去胺,碱洗(通常用饱和NaHCO3或10%NaOH) 可以除去酸性杂质。大多数情况下,当杂质既非酸性又非碱性时,可用蒸馏水洗涤, 以除去各种无机杂质。(注意:在摇动分液漏斗中的混合液体时,记住要经常排气,? 排气时使分液漏斗上沿口朝下,然后上举,在防护罩后面打开活塞。这样可以释放在 摇动液体时产生的气体压力。此外,在分液漏斗中放出液体之前,记住首先应打开盖 子。)
5)反向萃取回收损失的产品。如果你的产物有水溶性(含有几个极性基团),你可能需要用乙醚或乙酸乙酯反向萃取水层,以避免过多产物流失在水相中。可以使用TLC检测是否所有产物已经从水相中被萃取出。
6)在结束阶段进行盐洗(饱和NaCl溶液)。此操作有利于干扰乳化,并且可以除去溶于有机相中的水,起到“干燥”有机层的作用。
7)干燥有机层。将有机溶液和水相分离之后,在有机相中加入干燥剂以除去微量的水。通常用高效快速的MgSO4,但MgSO4有轻微的酸性;或用Na2SO4,它的干燥速度稍慢, 效率较低,但Na2SO4为中性。这些化合物可以和残留在有机溶液中的水结合,作用后形成团块。加入的干燥剂要适量,只要有一些干燥剂不再结块,说明可以不用再加入干燥剂了。(操作过几次后,你便会很好地掌握了。)
8)在干燥有机相时,可以将滤纸折叠好。参见Zubrick第136-138页。有些实验者喜欢在轻微减压下用布氏漏斗和未折叠的滤纸(或多孔漏斗)作为标准的过滤方法,目的是为了得到更高的产率。
9) 用叠好的滤纸和大漏斗(或布氏漏斗)将溶液滤入大的圆底烧瓶。为了防止在旋转蒸发时爆沸,溶液量不要超过圆底烧瓶容量的一半。
10) 旋转蒸发浓缩溶液,然后将产物溶解在少量溶剂中,并将其转入一个稍小的已知重量的圆底烧瓶中。
11) 再次旋转蒸发浓缩溶液。通过浓缩、加入二氯甲烷,然后重复几次操作,高沸点的溶剂可被有效地除去。
12)用真空泵除去残留的溶剂。对于非挥发性的化合物,可以用真空泵高效地除去残留的溶剂。这儿有一个加快此过程的窍门:排空圆底烧瓶,充入氮气,重复此过程, 然后用真空泵抽30分钟。如果你的产物是挥发性的(低分子量和/或低沸点),应该用旋转蒸发仪而不是真空泵抽至样品恒重。
13)样品恒重。从真空泵(或旋转蒸发仪)上取下圆底烧瓶,称重,然后继续蒸发15
到30分钟,再次称重。一旦连续两次得到的重量一样,你就可以准备去做NMR测定了。
8.5. 无氧操作指南
真空箱的操作:
参见LLP第9.2节有关使用对空气敏感的试剂和如何使用两通道真空箱的详细讨论。
溶剂脱气:
脱除溶剂中水和氧气的最好办法是在合适的干燥剂(比如金属钠)处理后进行蒸馏。有时,精馏操作可能比较烦杂(还可能比较危险)。由于在5.301中我们仅需要用到少量的无氧溶剂,因此采用惰性气体(如N2)来吹洗溶剂是更为有效的方法。
1)在一个热的圆底烧瓶中加入活性分子筛,用氮气吹洗直至降至室温。由于玻璃很容易吸附空气中的水气,因此必须在烘箱中(或火焰上)使所有的玻璃仪器彻底干燥。分子筛就像海绵一样不断吸附溶在溶剂中的水分。
2)当圆底烧瓶冷至室温后,加入溶剂并用橡皮膜封住瓶口,再用铜线把橡皮膜扎紧。
3)将一只干净的针头刺透橡皮膜,直接伸入溶剂中,鼓入气体。用另一只针头,使圆底烧瓶通大气。你将看到气泡产生。
4)持续15~20分钟足够了。
另一种溶液脱气的方法是“冰冻-抽气-解冻”法。先使溶液冰冻成固体(比如使用液氮),再用真空泵抽真空几分钟,然后关闭真空系统,使溶液缓慢升至室温。重复此操作至少两次以上。注意:有的极性溶剂,比如水、甲醇和乙腈在凝固时会膨胀,有可能引起玻璃容器破裂。在5.301中,我们不采用这种方法脱气,但你在将来的实验中可能会遇上。
导管转移:
参见LLP第6.4节。
过滤溶液:
过滤装置由三部分组成:装有样品的施兰克(Schlenk)瓶、施兰克接头和接收用的施兰克瓶。
1)趁热在玻璃仪器接头的两端涂上油脂。把接头的一端接上接收瓶(别忘了加入搅拌棒!),另一端接上另一只14/20开口的小烧瓶,用Keck夹子夹紧。抽真空,充入氮气(注意不要将油从鼓泡器中吸入真空箱中)。重复抽真空-充入氮气至少三次。
2)当你准备开始过滤时,将样品瓶及接受瓶中都处于正压的氮气流的氛围中。迅速将作为盖子用的小瓶从接头处移走,同时将样品瓶上的隔膜迅速移去,然后连接两部分,并把装置倒转过来。
3)关闭原料瓶的氮气,并且在接收瓶内稍微抽真空(注意在真空系统上装上冷却装置,以防止溶剂蒸发破坏贵重的真空泵!)。然后,可将真空泵关闭,因为在微小真空度下过滤可以顺利进行。
4)当溶液转移结束后,在接收瓶中充入氮气,将接收瓶中接头取下,再用橡皮隔膜封住瓶口。
8.6. 双溶剂重结晶指南
综述:
对于双溶剂重结晶,其中的一种溶剂(#1溶剂)应能使你的目标化合物在溶剂沸点时完全溶解;另一种溶剂(#2溶剂)在加入到目标化合物在第一种溶剂的饱和溶液中时,能够诱导该化合物结晶。
参考资料:
参见Zubrick第114-117页。
重结晶步骤:
1) 第一步是过滤除去不溶性杂质。
2) 将原料转入一只装有搅拌子的50mL锥形瓶中。加入过量的#1溶剂(在实验3.1中, 约加20mL),然后磁力搅拌加热至沸腾。用过量的溶剂是为了防止目标化合物在过滤过程中沉淀析出。
3) 在预热好的漏斗中,通过折叠滤纸过滤除去不溶性杂质(在过滤前,先用热溶剂预热漏斗,以防原料残留在滤纸上造成损失)。
4) 用2mL热溶剂洗涤锥形瓶和滤纸。
5) 蒸发除去过量溶剂,使溶液体积缩减至约15mL。
6) 将溶液冷至室温,此时溶液可能还不是饱和溶液,晶体可能不会析出。
7) 逐滴加入#2溶剂,直到溶液刚出现浑浊。再次加热溶液至沸腾(注意需搅拌!),继续加入#2溶剂。每加入一滴#2溶剂,你会观察到出现浑浊又溶解的情况,一直加#
2溶剂到溶液饱和(如果再多加一滴#2溶剂,浑浊将不再消散,此时溶液达到过饱和)。如果达到这样的状态,加入一滴#1溶剂使溶液再次澄清。
8) 将烧瓶从热源上移开,用磁铁将搅拌子取出,静置使之冷至室温,再放入冰水浴中。
9) 冷却一份双组分溶剂(其比例与配制上述饱和溶液时所用混合溶剂的比例相同)。此冷却溶剂将用于晶体的洗涤。
10) 在小号布氏漏斗上减压抽滤晶体,并用冷混合溶剂洗涤。
11) 滤饼先用空气吹干,然后置于真空下彻底干燥,称重并计算产率。干燥产物的办法之一是将产物放入一个预先称重的小瓶中,然后将小瓶放入真空干燥器中。你可以用面巾纸封住瓶口并用橡皮筋扎紧。
8.7. 培养单晶指南
综述:
你将会发现,培养单晶不仅需要耐心,而且还需要一双灵巧的双手。结晶过程对温度和其它轻微的扰动都非常敏感。因此,你应该在相似的条件下多尝试几个不同的实验温度,并为单晶的生长寻找一个没有干扰的安静环境。这里有一些经验性的贴士供你参考,以利于你的实验开展。
方案#1
● 有时好的单晶仅需冷却溶液即可生长。你也可以尝试加热溶液至所有物质完全溶解, 达到过饱和,再慢慢地使其冷却。
方案#2
1)选取一种可以溶解你的目标化合物的溶剂,制成饱和溶液。
2)如果有必要,可以通过过滤除去其中的不溶性杂质。对于少量溶液,可使用一种有效的过滤器,其制备方法是:将玻璃毛(甚至可以用面巾纸)塞入一根一次性Pasture滴管中,然后填入一英寸左右助滤物(如硅藻土Celite)。用新鲜溶剂湿润硅藻土,然后用球形压力器将溶液压过该管进行过滤。
3)寻找另一种溶剂,使目标化合物在其中不溶解(或仅微量溶解),而且这种溶剂能够和前一种溶剂混溶,并具有较低的密度。
4)将第二种溶剂小心地铺在小瓶中饱和溶液的上面。在两相界面上可看到一些混浊物。单晶将会沿着这个界面生长。
方案#3
● 将盛有饱和溶液的小瓶放置在另外一个较大的瓶中。在外面的大瓶中加入第二种溶剂并且盖紧盖子。第二种溶剂将会慢慢地扩散到饱和溶液中,晶体就会出现了!为了进一步减慢这个过程,可将这个扩散装置放在冰箱中。
可以尝试的溶剂系统:
CH2Cl2/乙醚或戊烷....................................THF/乙醚或戊烷
甲苯/乙醚或戊烷.......................................? 水/甲醇
CHCl3/正庚烷
8.8. 蒸馏操作指南
综述:
蒸馏在提纯试剂及分离粗产品时非常有用。蒸馏可分为两类:常压蒸馏和减压蒸馏。常压蒸馏操作比较简单,而减压蒸馏涉及一些较复杂的技术。两种方法在5.301中都要用到。
玻璃仪器:
蒸馏需用到一些专用于这一技术的特殊玻璃仪器。蒸馏装置有多种类型,但我们只用到其中的两种。在这两种装置中,都要用到一种短程蒸馏头,两者不同之处在于维格勒
(Vigreux)柱的使用。尽管在5.301中我们将不用其他类型装置,但你应该通过阅读熟悉他们。
常压蒸馏操作步骤:
1)收集必要的玻璃仪器:短程蒸馏头,温度计及温度计接头,接收瓶(至少两只), 维格勒蒸馏柱(可根据具体情况选择—参见LLP第196页)。
2)预热油浴或加热套。如果蒸馏物的沸点未知,此步骤应该略去。记住,多数情况下, 热源的温度需比蒸馏物的沸点高20~30°C。注意:由于热分解及可能着火,只在加热? 温度低于200°C时使用油浴。
3)记录贴有标签的接收瓶的重量。
4)将要蒸馏的物料放入带搅拌子的圆底烧瓶(搅拌子用于防止爆沸)。选择圆底烧瓶的大小非常重要。液体装至瓶子溶剂的1/2到2/3为好,液面太高将过早沸腾,液面过低则要花费太长的时间来蒸馏。
5)装配玻璃仪器,确保所有接口密闭性良好。如果拿不准要用多少夹子,记住组装一套玻璃仪器应至少使用两个夹子。对于常压蒸馏,不需要用油脂来密封接口。(注意:对空气或水敏感的化合物,蒸馏装置应用加热法干燥过,并在氮气或氩气保护下蒸馏。在5.301中,我们不进行此项操作,但在UROP中可能会遇到。)
6)蒸馏柱的保温。当用维格勒柱时,柱子应该用玻璃棉或铝箔来包裹。如果不进行隔热保温处理,蒸馏时要花费很长的时间。
7)将冷凝管连上水管,打开水龙头,检漏。
8)升起搅拌台及加热装置使之与圆底烧瓶接触,开始加热。注意:调压器的刻度表与?温度并一一不对应。将刻度表设置在70并不意味着将油浴加热到70°C,事实上,通常会升到更高的温度。另外,不同的油浴或加热套在相同的电压下得到的温度也不同。
9)放下通风橱挡板。这样可以避免意外伤害,同时也可以使蒸馏装置不受实验室空调的影响。空调将使蒸馏装置温度降低,并延长蒸馏时间。
10)不要加热过快!!!耐心是蒸馏成功的关键。
11)缓慢升高加热器的温度,直到溶液开始回流。
12)等待并观察蒸馏温度计的变化。如果10分钟后观察不到温度变化,则应稍微调高?温度。
13)重复步骤12,直到能观察到温度计有变化。一旦有变化,即准备收集馏分。
14)使蒸馏装置保持恒定的温度。使记录的蒸馏温度的至多在5°C范围内波动。
15)收集馏分直至温度发生突变。通常,当一种馏分蒸馏完成时,蒸馏温度计显示的温度将下降。此时,你应该更换接收瓶,或完全停止蒸馏。
16)当你已经收集到所有需要的产品,关掉加热电源,并让整个装置冷却下来。
17)称量接收瓶的重量,得到产物重量。
减压蒸馏的步骤:
1)收集玻璃仪器:与常压蒸馏相同,不同之处在于减压蒸馏需要用一只3口或4口转接头。另一很有用的玻璃仪器是Perkin三角器,但我们在5.301中没有用到它。这种仪器在课本中有介绍,在你以后的化学事业中将很有用。
2)按常压蒸馏的2~4步操作。
3)装配所有玻璃仪器,确保在所有接头上涂上油脂。注意节约真空油脂,它比较贵, 同时你也不想让它进入你的产品中吧。参见:Zubrick第53-55页,关于接口真空油脂的相关讨论。
4)按常压蒸馏的6~7步操作。
5)不要开始加热!!!
6)缓慢地将蒸馏装置抽真空。你应该可以看到液体开始起泡。不要担心,一切正常。在室温和减压条件下,残留的溶剂及低沸点的杂质将很快被蒸走。(这是一个说明为什么要将冷阱放在液氮中的很好的例子,否则这些化合物将直接进入泵油中!)
7)一旦泡沫减少,或减慢到几乎停止,你就可以开始加热了。
8)按常压蒸馏的9~15步操作。
9)卸去真空。当你已经收集到所需产品时,还不能将加热装置降温。首先,你必须卸去真空。但在做此之前,需确保所有接收瓶都用夹子、接口夹或你的手等方法固定在装置上。你不想看到在卸去真空后产品接收瓶摔得粉碎吧!如果一切准备就绪,向装置中通入氮气,然后移走热源,并让装置冷至室温。
10)所有物品都冷却后,称量接收瓶,计算产物的重量。
8.9. 快速柱色谱使用指南
综述:
快速柱色谱(Flash Column Chromatography)是一种快速而且(通常是)容易的分离复杂混合物的方法。在5.301中,我们要进行相对较大量的快速柱色谱分离,分离约1g左右的物质。色谱柱通常都比这根柱子小,你们中的一些人在进入研究室后会有这方面的实践体会。柱色谱和薄层色谱的原理一样,但它可以用于制备量物质的分离。因为我们是用压缩空气将溶剂推过柱子,故称其为快速柱色谱。这不仅使分离效果更好,并且可缩短过柱时间。
读物:
更加全面的描述,请参阅LLP第205-214页。
制备和操作快速柱色谱:
1)确定干燥、不含溶剂的待分离混合物的重量。
2)用薄层色谱选取溶剂体系,使Rf的值处于0.2~0.3之间,但如果混合物复杂,这可能不现实。在比较复杂的情况下,可能需要借助梯度洗脱的方式,简单地说,就是在纯化洗脱的过程中不断提高溶剂的极性,该技术在后面有更加详尽的介绍。但是在薄层色谱分析中,你必需确定哪种溶剂体系将会使不同的点样处于Rf在0.2~0.3的范围之内。
3)确定用于样品上柱的方法。你可有三种选择:净试样法,溶液法或硅胶吸附法。净试样法:如果样品是非粘性油状物,使用净试样法最为容易。你可以用一个长的
滴管过滤器将液体引入柱中,然后用预先确定的溶剂体系进行淋洗,把所有组分洗入柱子中。
溶液法:净试样法有时可能会引起分离柱断层。因此,对于液体和固体,更为普遍的方法是将样品溶于溶剂中,然后将溶液加入分离柱。最理想的状态是,混合物中所有组分在该溶剂体系(通常是戊烷或己烷)中的Rf为0。这在多数情况下是难以实现的,所以可选用那种只移动混合物中一个化合物的溶剂,或者你可以简单地用所选择的洗脱液。记住:后面两种选择对于难度较大的分离纯化是有风险的。
硅胶吸附法:最后一个技术是将化合物沉积(吸附)到硅胶上,这对部分液体和所有固体都是有用的。注意:硅胶是酸性的,因此这一步骤将会破坏一些对酸敏感的化合
物,它们通常需要在硅胶柱上再生。首先,在一圆底烧瓶中将混合物溶解在二氯甲烷中,加入硅胶(硅胶的质量大约是化合物质量的两倍)。在旋转蒸发仪上浓缩该溶液。注意:?硅胶是非常细的粉末,很容易被吸入旋转蒸发仪中。用玻璃毛塞住接头或泵的保护装置,以防止固体被吸入泵中。快速转动亦可以避免这个问题的出现。当固体基本上干燥的时候(当多数固体从容器壁上脱落,说明固体已经干燥),从旋转蒸发仪上卸下烧瓶,再用真空泵将溶剂抽尽(假设混合物中没有易挥发性物质)。注意:用玻璃毛塞住真空泵?接头,否则你可能会发现硅胶(以及你的化合物)进入真空管并沉积在那里。一旦其完全干燥之后(固体中再没有气泡产生),从真空系统中取下烧瓶,用干净的刮刀从壁上刮下固体。现在,你可以简单地用粉末漏斗将这部份固体加到分离柱的顶端,然后用洗脱液淋洗(每次1.5mL)。
4)确定合适的硅胶和化合物的比例。对于简单的分离,通常要求两者的比例为30~50:
1(重量比);但对比较困难的分离,需要的比例高达120:1。阅读LLP上的相关材料,并且和一些经验丰富的同事进行讨论,将对你解决这个难题大有裨益。
5)选取合适的分离柱。你所需用的硅胶量决定了分离柱的尺寸。是使用短而粗还是长而细的分离柱,迄今为止还没有定论。在5.301中,我们认为短而粗的分离柱会有更好的分离效果,但是这个结论可能会被你以后的一些同事所质疑。当你首次开始实验的时候,最好的选柱方法是向实验室的同事了解:对给定的硅胶量,应选择怎样的分离柱! 把结论记在笔记本上(这比测量分离柱的直径方便很多)。在5.301中,我们只用一种型号的柱子,这就不存在什么选择问题了!
6)选取合适的收集用试管。这也是一个向有经验的同事们请教的好机会。但也有简单的方法:将硅胶体积除以4,然后选取能装下这个体积的试管就可以了。(200mL的硅
胶对应于50mL的组分)
7)一旦你选定了分离柱,你需要堵住活塞底端以避免硅胶的流失。通常,用一小团棉花或者玻璃毛加一根长棍或玻璃棒即可完成。
8)在通风橱中填充分离柱。考虑到要用大量挥发性溶剂,以及干燥硅胶对于健康的危害,不允许在通风橱外进行柱的操作。检查并确定柱子是否完全垂直,倾斜的柱子不利于分离。
9)关上活塞并且加上几英寸高的洗脱液。
10)用漏斗向分离柱中加入一些沙(干燥并且经过洗涤的)。目的是在塞堵物上铺一薄层砂(不超过1cm),这样可以避免硅胶落入收集瓶中。
11)量取合适量的硅胶,最安全的方式是在通风橱中量取。硅胶的密度大约是0.5 g/mL,因此可以直接用锥形瓶量取(100g=200mL)。不要让硅胶的体积超过烧瓶的1/3,因为我们还要在其中加入溶剂。
12)在刚量取的硅胶中加入至少1.5倍体积的溶剂,将其制成浆状,用力振荡和强烈搅拌,使其充分混合,并且除去硅胶中的气体(气泡的存在将会使分离柱的效率大打折扣)。
13)用粉末漏斗小心缓慢地将浆状物移入分离柱中,注意不要破坏下面的沙层。注意在灌浆的过程中不时地停下来并且摇动浆体,以确保硅胶混合均匀。灌浆结束后,用洗脱液反复冲洗烧瓶几次,并且将余下的溶剂硅胶混合物加入到分离柱中。
14) 用滴管和洗脱液将黏附在柱子顶部边缘上的硅胶冲洗到溶剂层中。
15)当所有的硅胶都被洗离柱壁,打开活塞,用压缩空气给柱加压。柱内的硅胶将会压缩到原来高度的一半左右。检查以确保柱子的顶段平坦,如果不平,必需重新搅拌,然
后沉降下来。在加压下,加入过量的洗脱液,用铅笔头或橡皮塞轻轻地从敲打柱子,这?将使硅胶颗粒填充得更加紧密。收集从柱子中流出的所有洗脱液,在加入化合物之后重复使用。
注意:切记不要让溶剂液面低于填充层。
16)当柱子填充好以后,在硅胶的顶部加入沙子作为保护层。沙层需要填充的比较平整,厚度约在2cm左右。这在添加溶剂时起到保护柱子的作用——当溶剂加入过快时,如果没有沙层的保护,溶剂可能会破坏填充硅胶的平整的表面(因此影响分离效果)。
17)在溶剂还没有达到沙层之前,可以用压缩空气促使溶液层下降。
18)关闭活塞,将第一个试管放在柱子的出口下面。
19)小心地向分离柱中加入你的化合物——当添加液体时,确保是沿柱子的壁面加入,而不要直接滴加在柱的顶段。当冲洗含有混合物的烧瓶时,小心地一次性地将满满一滴管淋洗液加到分离柱中。然后打开活塞,当液体下降到填充物的顶段时关掉活塞。如此冲洗烧瓶三次。对沉积在硅胶上的混合物,还要再加2cm厚的保护沙层。
20)小心地在分离柱中加满洗脱液。开始时可以用巴斯德球管加入溶剂。当加入了1cm高度的溶剂之后,最好打开活塞。继续用滴管滴加溶剂,直到溶剂高于柱内的填充层几个厘米。现在,可以通过一个粉末漏斗从锥形瓶中加入溶剂了——缓慢地让它沿柱子壁加入。一定要有耐心,不要破坏柱内填充物的顶段。
21)当把洗脱液装满分离柱之后,你就可以开始“过柱”了。请记住快的流速将使分离更好地进行。调整空气压力使达到一个快的流速——但不能象消防笼头那么快!保持压力,在收集试管装满后换上一支新的试管。注意随时向柱内补充溶剂。
22)用TCL跟踪柱子的分离进程。一边收集样品,一边进行薄层色谱分析。这一点可能带来一些忙乱,因此,你若要观察色谱柱工作进展情况,则在一开始便可以减低气压
(甚至完全去除)。
23)当操作梯度洗脱时,先用一种溶剂以保证具有较大Rf的化合物先从柱中被洗脱。当他们被安全地洗脱到收集烧瓶之后,便可以更换一种极性更大的溶剂继续洗脱。注意:?逐步提高溶剂的极性。过于急速的极性变换可能会使硅胶分裂——就像电影里可怕的地?震场面一样,柱内的填充层出现裂缝。这对你的分离将会非常不利!因此,以每100mL
(或更多)溶剂中增加5%左右的极性,直至达到所希望的溶剂。然后,用该种洗脱液洗脱,直到目标化合物被洗脱出来。这个时候,你可以继续更换洗脱液或者直接进行下一步。
24)当你确定所有目标化合物都已经从柱内被洗脱的时候,你就可以将每样东西收拾起来了。首先,在分离柱的底端放置一个大烧瓶,然后用一个夹子切断联通分离柱的压缩气体的气路。让气体将剩余的溶剂全部压出柱子,然后干燥硅胶(从分离柱中移出硅胶比较困难,除非它是完全干燥的)。对于大的分离柱,这个过程差不多要耗费一个小时。
25)当分离柱在干燥之时,可开始将组分合并起来。用薄层色谱来确定哪个试管中含有所需的纯样品。将相似纯度的组分合并放在大的圆底烧瓶中,并用旋转蒸发仪进行浓缩。对于费时较长的柱子,可在柱分离过程中就合并流出的组分,以加速进程。
26)当完全除去溶剂后,就可以用NMR分析所得到的化合物了。
9.1. NMR的操作指南
Varian Mercury 300 Plus FT型核磁共振(NMR)波谱仪的操作
(仪器旁有一套完整的操作说明)
核磁共振(NMR)波谱仪是你在课程5.301中用到的最复杂(也最昂贵)的仪器,因此,花一些时间来熟悉并正确掌握仪器操作是非常重要的。本指南包括了一个简单的1H NMR谱所需的操作步骤。但是,你应该注意到其中的步骤2~7将被略去。在1月7日星期二那天,要求你在助教的帮助下测得第一张1HNMR谱。那时,助教会向你演示该仪器的正确操作方法, 同时有机会让你做详细的笔记,并让你询问做什么和为什么要做。在IAP将结束的时候,你应该对做1HNMR谱图比较熟练。在足够多的练习后,你最终能够在10分钟之内完成下列的7个操作步骤后而得到一张1H NMR谱:
1) 放置试样;........................?5)处理数据;
2) 锁场;....................................6)积分;
3) 匀场;....................................7)标图,结束实验。
4) 获得谱图的FID;
1) 放置试样:
● 用面巾纸擦拭干净装有你的试样的带帽的NMR管。
● 用所提供的测深仪将NMR管插入仪器的悬标中。
● 再次清洁NMR管。
●点击[eject],你必须听到喷出气体的声音才能继续实验。如果没有听到声音,需要检查液氮罐的阀门。
●将NMR管和悬标放在磁体顶部的管孔中,并且点击[insert],样品将下落并进入磁场中。
● 等听到滴答一声的时候,点击[on]开始。
● 记录:
2) 锁场:
锁场的目的:在1H NMR谱图中,质子频率的差异是ppm级甚至更小,因此累加的各FID值必须相当一致。磁场强度的轻微偏移将会产生很大的问题, 因此仪器必须保持一个稳定频率作为参考,以此修正各个FID值,可以保证正确累加。这就是氘代锁场的作用。氘代溶剂中氘原子的振动频率与试样分子中质子完全不同,他们只有一个信号峰。因此仪器跟踪氘原子的信号,对1HNMR信号做相应的调整,这样磁场的任何漂移都会被补偿。
● 说明:
3) 匀场:
匀场的目的:“shim”这个词过去用于描述调音师通过打磨处理铃的表面,使铃只发出一个强的音调的铃声。我们的目标是通过降低(或增加)局部的磁场,使整个样品中磁场强度处处均匀。否则,由于分子中相同质子在不同频率时振动(取决于核磁管在不同强度的磁场中具体所处的位置)使谱图出现宽峰。将核磁管处于20转/秒的转速是模拟其在XY面上各向同性的一种方法。
我们将在Z轴向上进行匀场,即调整垂直方向上的磁场。通常,我们总是尽力得到响应最大的锁定信号。除了溶剂,决定匀场是否容易的主要因素是样品的高度。如果溶剂过少
(高度小于4cm),你的匀场过程将会非常麻烦,因为空气/溶剂界面靠近产生NMR信号的磁场线圈。为了使匀场简便易行,好的匀场程序最近已经输入电脑,你只需要调用他们就可以了。
● 说明:
4) 获得谱图的FID(自由诱导衰减):
● 说明:
6) 积分:
你可以对NMR中峰进行精确的积分,从而判断每种质子的相对数目。
● 说明:
7) 标图,结束实验:
● 说明:
9.2. 红外(IR)操作指南
本指南贴在本课程5.301中所使用的FT-IR旁边的墙上。
背景扫描:
首先,按仪器背后的凸起的按钮,使屏幕升起来。再将背景卡插入到分析槽中并输入如下的命令:
● 背景
● 扫描
● 扫描次数——通常是四次
扫描结束后,将会出现一张谱图,现在你可以将背景卡换成待测样品。
样品扫描:
(扫描结果将被存储到X,Y或Z缓冲器中。)
输入如下的命令:
● X,Y,或Z(选取缓冲器,储存你的谱图,同时将覆盖原来缓冲器中的所有信息)
● 扫描
● 扫描次数——通常是四次
准备绘图:
● 打开仪器左侧后边的开关,开启绘图仪;
● 将两支笔放入绘图仪中;
● 将纸调整到合适的位置——将其移至左侧,并触及上面的白色标志;
● 用左侧小的黑色手柄将纸锁定;
● 启动IR控制台上的“Plot”键;
● 等待绘图完毕;
● 取出图并盖好笔;
● 关掉绘图仪。
9.3. 气相色谱(GC)使用指南
概述:
在课程5.301中,我们将用到一台非常精密的HP5890 Series II型的气相色谱仪,配有HP 3365工作站控制的HP 7673自动进样器和HP 18765条码识读装置。这意味着你将用到一台重要的仪器,而操作却非常简单。
为自动进样器准备样品:
与通常的研究实验室不同,在课程5.301中,你不必将样品手动注入GC柱中,你可以用自动进样器来完成所有需要的进样工作。因此,你所有需要做的工作仅仅就是把待分析的样品移入到特制的自动进样瓶中。
1) 用一个特制的分配器,在每一个自动进样瓶上贴上条形码(在你的笔记本上记录这个号码,这样你就不会搞不清哪个号码对应哪个样品了)。检查分配器上的图标,以确定瓶子正确排列,检查并确保整个标签都粘在瓶上。
2) 吸取1mL左右GC样品(参照GC样品制备指南准备试样),注入样品瓶中。样品的高度恰好在条形标识的下面是比较适宜的,避免过高或过低。
3) 用封口机将瓶子封上盖子,不要压得过重,中等压力就可以了。确保不能用手旋动盖子,并且在瓶子颈部没有褶皱,隔膜平整居中,如果有任何缺陷就不要用,否则进样器很容易受损。这时,可用剪钳去掉有缺陷的盖子,重新配置合适的盖子。
4) 所有进样瓶准备完毕后,向助教请教如何操作该仪器。
9.4. 紫外-可见(UV-Vis)分光光度计使用指南
用HP 8542双光束分光光度计测UV-Vis谱的说明
用于蛋白质鉴定实验:
1) 在HP 89532A软件的主菜单中,用程序选择器选择Kinetics这个选项。
2) 几个缺省的参数需要更改。选择Time/Cell,设定周期时间为2秒,运行时间为6秒;随后选择Functions选项;然后选择单波长。根据你做哪个实验的需要,输入596或562。
3) 在比色皿中注入去离子水,作空白扫描,提供光谱背景。注意:不要触及比色皿的?光洁面,否则会干扰光路。仔细地用擦镜纸把比色皿外表擦干净并保持干燥,将充满水的比色皿放于管架中,推动黑色的拉杆将其固定在设定位置。分别选择Scan Screen,Pre-Run,然后是Meas. Blank,大约5秒钟后,空白扫描结果出现,是一条噪声很大的线。不用担心,这是正常的,是电脑自动处理的结果。
4) 打开样品架,取出空白溶液的比色皿,放入装有第一个试样的树脂比色皿。选择Start
Run,然后选择Begin,这样仪器就会分四次测量样品的吸光度值。
5) 若要进行吸光度的硬拷贝,需要进行如下的操作:选取Return,Tabulate?和Time
Traces。这样就可以打印出所有的吸光度值。通常,建议你对同一个样品进行多次的扫描测量,以检测仪器的精确度。你可以将比色皿快速地从池架上取下来,用擦镜纸将其擦拭干净,将其放回原来的位置,然后进行另一次扫描。如果本次测量的结果和第一次所得到的值差异较大,则多测量几次,直到至少有3次测量数据相近。
6) 一旦你的测量数据打印出来,即进行下一次扫描,请按Escape键,然后选取Scan
Screen。
7) 当你对某个样品的实验数据表示满意,则可开始对余下的样品重复步骤4~6进行测量。
手机版